|
A last type of effect of misfolded protein is seen in amyloid diseases; in which protein deposits in the cells as fibrils. A number of common diseases of old age, such as Alzheimer's disease fit into this category, and in some cases an inherited version occurs, which has enabled study of the defective protein. One such example is transthyretin, a protein that transports thyroxin and retinol binding protein in the bloodstream and cerebrospinal fluid. In senile systemic amyloidosis, which affects people over 80, transthyretin forms fibrillar deposits in the heart. which leads to congestive heart failure. An inherited version, Familial amyloid polyneuropathy (FAP) affects much younger people; causing protein deposits in the heart, and in many other tissues; deposits around nerves can lead to paralysis. FAP patients are mostly heteozygous for the transthyretin gene, and over 50 single site mutations are known. The structure of wild type TTR was determined in the 1970's by Colin.Blake and his colleagues; the biological unit is a tetramer. Each monomer has two 4- stranded b-sheets, and a short a -helix. Anti-parallel b -sheet interactions link monomers into dimers and a short loop from each monomer forms the main dimer-dimer interaction. These pairs of loops keep the two halves of the structure apart forming an internal channel. Here are two views of the tetramer. Side view
 Top view  The internal channel has two binding sites for thyroxin.
Only a small proportion of the plasma TTR molecules are involved in
thyroxin transport, a much higher proportion are found tightly bound to
retinol-binding protein (RBP); the function of this is thought to be to
stabilize the binding of retinol to RBP, keeping it in the plasma by
reducing its filtration from the blood by the kidneys. Retinol is made
from Vitamin A, and is important in regulation of vertebrate
development. 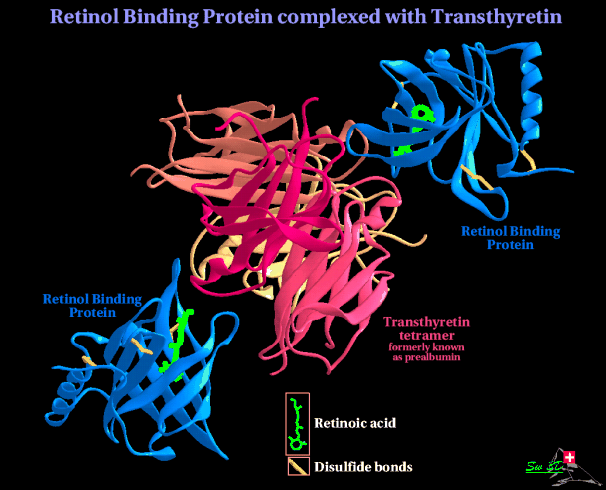 The structure of several FAP TTR variants is known, and
they are virtually identical to the normal form .To examine the stucture
of transthyretin and the mutants interactively the following links will
allow you to see the stuctures using Rasmol or Chime.
The pdb files from expasy can be seen with Rasmol if you have it
installed, and from the new PDB (Protein Data Bank) site at RCSB there are
several options available (click on view structure, and choose from VRML,
Chime, or Rasmol). The following links will allow you to see the original
wild-type stucture 2pab, from [expasy] or [RCSB], the later (refined) 1tta from [expasy] or [RCSB]; and the mutant forms 1ttb (Ala109 to Thr)
[expasy][RCSB] and 1ttc (Val30 to Met) [expasy][RCSB]. The PDB files of other mutants (such as 1tsh,1ttr), and
complexes of transthyretin with retinol binding protein, retinoic acid,
thyroxine and various inhibitors are also available from expasy,RCSB., or the PDB mirror
site at EBI.
Model of a helical b-sheet structure
Four twisted b -helices make up a proto-filament (50-60A), and four of these associate to form a fibril as seen in electron microscopy (130A). () Model of a fibril made up of 4 fibrils
Recently T. Klabunde and colleagues have studied the binding of thyroxine, and of some anti-inflammatory drugs, that inhibit fibril formation in vitro, to transthyretin. They showed that the binding site for thyroxine has 3 pairs of small depressions. Two of these on each monomer bind to iodines of thyroxine, in two alternative conformations, and the bound thyroxine stabilises the tetramer. They have designed some drugs that bind more strongly than thyroxine, which should therefore stabilise the tetrameric form, so there is now hope of effective treatment for SSA and FAP. (). |
 Index Page
Index Page